
Comment la mucoviscidose se manifeste ?
De premiers signes peu après la naissance
La mucoviscidose est une maladie génétique qui affecte plusieurs organes. Elle s’accompagne donc de symptômes divers, certains d’entre eux survenant souvent peu après la naissance :
- le méconium (ou « selles noires du nouveau-né ») n’est pas éliminé, ce qui engendre une occlusion intestinale avant même la première défécation ;
- la peau des enfants a un « goût salé » ;
- les bébés ont un poids insuffisant et souffrent de ballonnements ainsi que de douleurs abdominales; leurs selles sont grisâtres et grasses ;
- très tôt, les petits souffrent de toux chronique et de difficultés respiratoires.
Du mucus visqueux dans les voies respiratoires
Chez les personnes atteintes de mucoviscidose, les cellules productrices de mucus sécrètent un liquide très visqueux qui empêche le mécanisme de nettoyage des voies respiratoires de fonctionner. Les particules de saleté et les germes pathogènes qui y ont pénétré lors de l’inspiration ne peuvent alors plus être évacués. Il en résulte des infections respiratoires récurrentes et une toux chronique. À un stade avancé, la maladie engendre une destruction du tissu pulmonaire qui provoque une gêne respiratoire et un déficit en oxygène.
Perturbation de la digestion
Chez les personnes souffrant de mucoviscidose, le mucus visqueux bouche les canaux du pancréas, empêchant alors les enzymes digestives d’arriver jusqu’à l’intestin. Il en résulte une digestion insuffisante de la nourriture. C’est la raison pour laquelle les enfants concernés ont souvent des problèmes de croissance et sont plus tard en sous-poids.
Parmi les autres symptômes et maladies concomitantes observés à l’âge adulte, on recense une inflammation du foie et de la vésicule biliaire, du diabète et une ostéoporose (atrophie osseuse).
Causes et possibilités de traitement
Origine de la mucoviscidose – Causes
La mucoviscidose est provoquée par une anomalie génétique que les parents transmettent à leurs enfants. Des scientifiques ont localisé son emplacement exact en 1989: chez les personnes concernées, une information erronée contenue dans le chromosome 7 (sur les 23 paires de chromosomes que compte le corps humain) empêche une production correcte de la protéine CFTR (cystic fibrosis transmembrane conductance regulator). Or celle-ci est essentielle pour que le mucus et les sécrétions soient suffisamment liquides. En cas d’anomalie génétique, l’équilibre hydrique et salin du corps est perturbé: visqueux et collants, les fluides corporels bouchent des organes vitaux comme le pancréas, l’intestin et surtout les poumons.
Des enfants malades malgré des parents en bonne santé
En Suisse, près d’une personne sur vingt est porteuse de l’anomalie génétique sur le chromosome 7. Pourtant, seul un nouveau-né sur 3300 souffre de mucoviscidose. Pourquoi? Parce qu’un enfant ne souffrira de mucoviscidose que si ses deux parents portent la mutation dans leurs gènes et s’il hérite du gène anormal de chacun d’entre eux. Si l’enfant ne reçoit un gène défectueux que d’un des parents, il sera porteur de la mutation, mais en bonne santé.
Dépistage de la mucoviscidose – Diagnostic
Dépistage chez les nouveau-nés
Depuis 2011, un test est réalisé sur tous les nouveau-nés : au quatrième jour après la naissance, des spécialistes prélèvent quelques gouttes de sang au niveau du talon de l’enfant. Elles sont ensuite analysées dans le but de détecter dix maladies hormonales et métaboliques, dont la mucoviscidose.
Test de sudation
Si le dépistage réalisé chez les nouveau-nés montre une anomalie, un test de sudation peut être effectué pour confirmer le diagnostic. En effet, la sueur des nourrissons atteints de mucoviscidose présente une teneur élevée en sel. Chez 98% des personnes concernées, ce test indolore donne lieu à un résultat positif.
Test génétique
Ce test consiste à rechercher une anomalie génétique sur une goutte de sang ou un frottis de la muqueuse buccale.
Diagnostic prénatal
Dès les premiers mois de grossesse, on peut déterminer si l’enfant à naître souffre ou non de mucoviscidose. À cet effet, on prélève un échantillon de tissu du placenta («biopsie chorionique») entre la 8e et la 12e semaine de grossesse ou on procède à une ponction dans le liquide amniotique («amniocentèse») entre la 14e et la 16e semaine de grossesse.
Augmentation de l’espérance de vie – Traitement
Toutes les formes de traitement visent à préserver aussi longtemps que possible le fonctionnement des organes touchés. Grâce à de nouvelles possibilités thérapeutiques, l’espérance de vie et la qualité de vie des patientes et patients sont aujourd’hui nettement plus élevées qu’auparavant. Les nouveau-nés chez qui la maladie est identifiée lors du dépistage systématique ont de bonnes chances d’atteindre l’âge de la retraite.
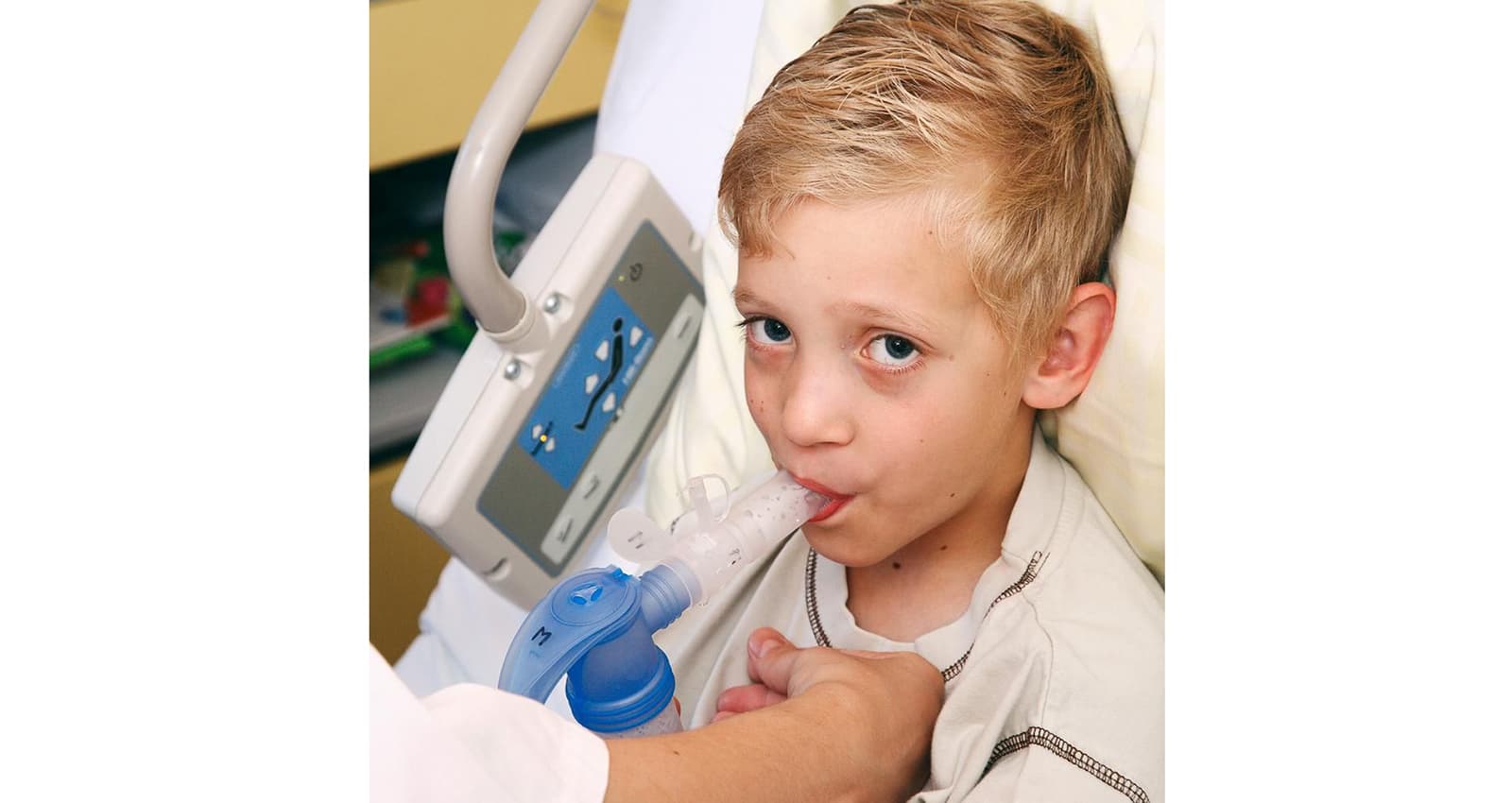
Une amélioration de la fonction pulmonaire grâce à une physiothérapie respiratoire
La physiothérapie accompagne les personnes concernées tout au long de leur vie. L’offre de prise en charge physiothérapeutique englobe en premier lieu la thérapie par inhalation et la physiothérapie respiratoire, mais aussi des exercices axés sur la force et l’endurance ainsi que des techniques de relaxation. Grâce à des exercices spécifiques, la physiothérapie respiratoire permet aux personnes concernées d’améliorer leur fonction pulmonaire et de parvenir à décoller le mucus visqueux de leurs bronches ainsi qu’à l’expectorer. Elles peuvent ainsi respirer plus facilement et sont plus résistantes face aux agents pathogènes.
Des antibiotiques contre les infections bactériennes
De fréquentes infections des voies respiratoires contribuent de façon décisive à la destruction des poumons des personnes atteintes de mucoviscidose. Cela est souvent dû à des bactéries que l’on peut garder sous contrôle au moyen d’antibiotiques. Un recours fréquent aux antibiotiques peut toutefois provoquer des effets secondaires tels que des allergies ou des résistances.
Rétablir le fonctionnement du canal chlorure
De nouveaux médicaments appelés « modulateurs de CFTR » (cystic fibrosis transmembrane conductance regulator) permettent depuis peu d’améliorer ou de rétablir le fonctionnement de la protéine CFTR (canal chlorure), et ainsi de réduire voire de stopper complètement de nombreux traitements symptomatiques.
Plus de calories et d’enzymes digestives
En raison d’infections récurrentes et du dysfonctionnement du pancréas, les personnes concernées souffrent souvent de malnutrition. Elles ont donc besoin de plus de calories et de vitamines que les personnes en bonne santé. Pour permettre à leur organisme d’assimiler les nutriments, elles doivent prendre des enzymes digestives avant et pendant les repas.
Transplantation pulmonaire
Si, malgré le traitement intensif, la destruction des poumons est trop avancée, une greffe de poumon peut être envisagée. Cette mesure est indiquée si l’espérance de vie estimée est inférieure à deux ans, si la fonction pulmonaire est fortement restreinte et si de graves infections surviennent fréquemment. La décision de procéder ou non à une greffe dépend de nombreux facteurs.
Si une telle opération est réalisée dans le cas d’une mucoviscidose, les deux poumons sont remplacés. Il peut toutefois en résulter des problèmes tels que des infections ou le rejet du nouvel organe.

Vous avez des questions ? Nous vous aidons !
Les Ligues pulmonaires cantonales offrent un conseil et un suivi complets. L'objectif est de permettre aux personnes concernées de vivre le plus possible sans symptômes. La Ligue pulmonaire cantonale la plus proche de chez vous répondra volontiers à vos questions.
